Drug information
Audio
| drug-audio-en-306.mp3 |
Pronounce:
Brand Name
Fuzeon
Other Names
T20
Drug Class
Fusion Inhibitor
Drug Image(s): (Click to enlarge)

Chemical Image: (Click to enlarge)
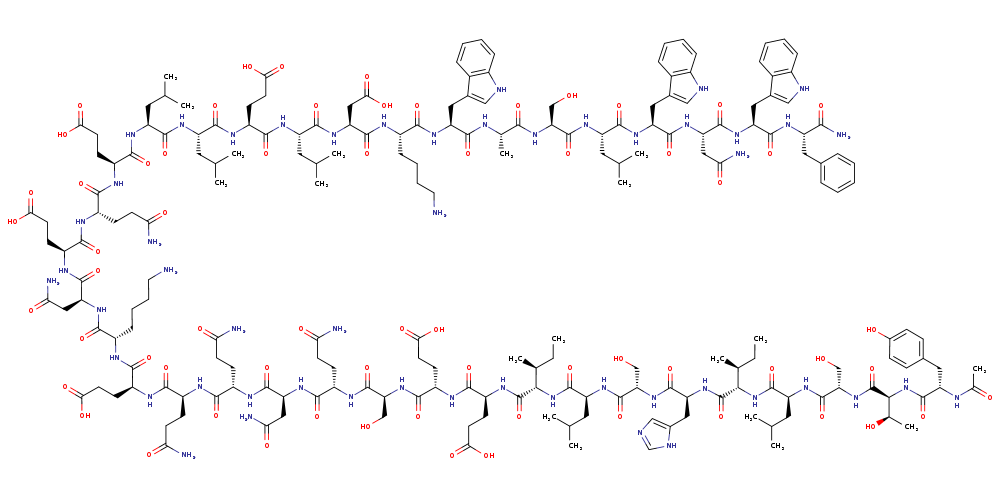
enfuvirtide
Molecular Weight: 4491.9149
(The drug image[s] shown above is of the brand product only. There may be other available products not shown.)
FDA label informationFDA label information
Several FDA-approved drug labels may be available for enfuvirtide. Clinicalinfo provides the following link to the DailyMed drug label solely as an example of the labels available for enfuvirtide. Inclusion or absence of a drug label link on the Clinicalinfo site does not imply endorsement or lack thereof by Clinicalinfo. Search DailyMed or Drugs@FDA to access more information on enfuvirtide, including additional drug labels and any generic equivalents.